Por ARNALDO SCHIZZI CAMBIAGHI
Histórico dos judeus Ashkenazi e Sefaradi
Qual a relação entre os judeus e a Reprodução Assistida?
Algumas doenças genéticas comuns aos judeus têm motivos originados na história do próprio povo e podem ser evitadas pelas técnicas de reprodução assistida. A história é antiga e começa aproximadamente a 450 anos AEC (antes da era comum), quando cerca de 40 mil judeus foram deportados para a Babilônia (hoje, Iraque), onde formaram comunidades e mantiveram suas práticas e costumes religiosos associados a outros costumes herdados dos babilônios.
Esse deslocamento é chamado de “primeira diáspora” (em grego antigo, διασπορά – “dispersão”), termo esse empregado para povos que são forçados ou incentivados a se mudarem para outras regiões. O termo “diáspora” é usado com muita frequência para fazer referência à dispersão do povo hebreu.
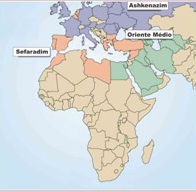
Muito tempo depois, em 70 DC, houve a segunda dispersão dos judeus (“segunda diáspora”), dessa vez para a Europa e o norte da África. As comunidades europeias se concentraram na França, Espanha, Roma e Alemanha. Os judeus da França e da Alemanha ficaram conhecidos como Ashkenazi (palavra hebraica para “alemão”). Os judeus da Espanha, que permaneceram sob o império árabe por centenas de anos, tinham conexão com os judeus do norte da África e do Oriente Médio, e acabaram sendo chamados de Sefaradi (palavra hebraica para “espanhol”).
Os Ashkenazi, no início, eram um povo “isolado”, isto é, um grupo populacional separado de outros, seja por barreiras geográficas, sociais, políticas etc. Devido a esse “isolamento”, os casamentos eram endogâmicos, ou seja, casamentos entre parentes próximos. Isso aumentou a probabilidade de homozigose (homozigose = o mesmo gene para um defeito repetido no par de cromossomos) entre os filhos resultantes dessas uniões consanguíneas.
Esse “estilo” de casamento fez com que determinados genes se tornassem prevalentes e a probabilidade de doenças recessivas graves fosse maior do que em outros grupos populacionais. Porém, ao longo dos séculos, muitos judeus Ashkenazi migraram para outros continentes, disseminando as doenças genéticas provenientes desses casamentos antigos e perdendo as características de um grupo isolado e parte de sua cultura.
Entretanto, o fato de a população judaica ficar isolada por muito tempo fez com que esses fenômenos genéticos ocorressem com mais frequência. Assim, ela apresentava algumas doenças mais prevalentes em relação a outras populações.
É importante que todos os judeus Ashkenazi que pretendem ter filhos façam um teste do DNA para as mutações das doenças autossômicas recessivas mais comuns para saberem se são portadores dos genes responsáveis. Algumas delas: Doença de Tay-Sachs (TSD), Fibrose Cística (CF), Doença de Canavan (CD), Disautonomia Familiar (FD), Síndrome de Bloom, Doença de Gaucher. As frequências de portadores e a incidência das doenças entre os judeus Ashkenazi foram calculadas em aproximadamente 1/31 e 1/3.900, respectivamente, para TSD, 1/25 e 1/2.500 para CF, 1/36 e 1/5.200
para CD, e 1/30 e 1/3.700 para FD. Outras doenças como Mucolipidose IV, Niemann-Pick tipo e a anemia de Fanconi grupo C são mais raras.
O Colégio Americano de Genética Médica (The American College of Obstetricians and Gynecologists) tem recomendado a triagem para a população judaica Ashkenazi, e o que poucos sabem é que a fertilização in vitro possibilita o diagnóstico pré-implantacional, evitando a transmissão para os filhos dos genes das doenças Tay-Sachs (TSD), Fibrose Cística (CF), Canavan (CD) e Disautonomia Familiar (FD).
Painel de triagem para as doenças mais frequentes em judeus Ashkenazi que podem ser realizados pelo IPGO

Os Judeus e reprodução assistida
O PGD (Pré-Implantation Genetic Diagnosis), que pode ser traduzido como DPI (Diagnóstico Pré-Implantacional), é um exame que pode ser utilizado no processo de FIV (fertilização in vitro) com o objetivo de diagnosticar nos embriões a existência de alguma doença genética ou cromossômica antes da implantação no útero da mãe. Assim, casais com chances de gerar filhos com problemas como Síndrome de Down, Distrofia Muscular, Hemofilia, entre outras anomalias genéticas, podem descobrir se o embrião possui tais doenças por meio desse exame.
Essa técnica, utilizada em tratamentos de fertilidade, consiste na retirada de uma ou mais células do embrião (biópsia embrionária), em laboratório, que são encaminhadas para análise antes mesmo de ele ser colocado no útero. Esse procedimento não afeta o futuro bebê. Indicamos o exame em casos de casais com risco de doença genética nos filhos por idade avançada ou antecedente familiar de alguma moléstia. Existem várias doenças que podem ser diagnosticadas por essa técnica, e o IPGO realiza esses tipos de exames, quando indicados.
Principais doenças genéticas autossômicas recessivas entre judeus Ashkenazi e as suas características clínicas
Doença de Tay Sachs(TSD)
A Doença de Tay-Sachs ou Gangliosidose GM2 é uma doença genética autossômica recessiva resultante de uma deficiência da enzima hexosaminidase A. Com isso, ocorre o acúmulo de uma substância chamada gangliosídeo GM2, principalmente no tecido neuronal, o que resulta numa degeneração neuronal progressiva.
A criança com a doença desenvolve-se normalmente até os 3 a 6 meses, quando então iniciam-se os sintomas lentamente, ocorrendo perda da visão periférica, regressão gradual das funções neurológicas e podendo o bebê se tornar incapaz de engatinhar, de se virar, sentar-se ou segurar coisas. Outros sintomas incluem aumento da perda da coordenação progressiva, incapacidade para engolir e dificuldades respiratórias. Eventualmente, a criança fica cega, com atraso mental, paralisia e não responde aos estímulos do ambiente que a rodeia. A Doença de Tay-Sachs (TSD) é uma doença grave, progressiva no sistema nervoso central, conduzindo à morte nos primeiros anos de vida. Atualmente, nenhum tratamento eficaz está disponível.
Hoje já se conhece o gene que determina a doença, o HEXA. Ele se localiza no cromossomo 15 (15q23-q24) e codifica a subunidade alfa da hexosaminidase A. Mutações nesse gene levam a 98% dos casos de Doença de Tay-Sachs na população judaica Ashkenazi. Como é uma doença autossômica recessiva, é necessário ter os dois genes mutados para que este seja o diagnóstico, portanto, tanto o pai como a mãe devem ser portadores heterozigotos do gene. A frequência de portadores das mutações que causam a doença de Tay-Sachs é extremamente aumentada na população de origem judaica, ocorrendo em cerca de um em cada 36 nascimentos em países ocidentais. A incidência da doença também é grande, atingindo aproximadamente um em cada 3.600 nascimentos nesta população. Um estudo realizado em Porto Alegre apresentou uma frequência de portadores de 2,4% para uma população judaica de origem Ashkenazi. A elucidação das mutações que causam a doença de Tay-Sachs introduziu a possibilidade de utilizar testes para detectar portadores das mutações na população geral e nas populações com maior prevalência da doença. O estudo é indicado para pessoas com história familiar de Tay-Sachs e deve ser feito principalmente em famílias de origem judaica, nas quais a incidência da doença é alta, para a detecção de portadores e auxílio no aconselhamento genético.
Síndrome de Bloom
É uma doença autossômica recessiva, que atinge o cromossomo 15 causando a alteração de um gene localizado em 15q27.1. É caracterizada por retardo no crescimento, predisposição para infecções, neoplasias malignas, lesões na pele causadas pelo sol (telangiectasias facial ou pigmentação anormal) e, em alguns casos, dificuldades de aprendizagem e retardo mental. A idade média de morte é com 27 anos e, geralmente, está relacionada ao câncer. Nenhum tratamento eficaz atualmente está disponível. Essa síndrome é rara em outras populações, mas acomete cerca de um em cada 100 judeus Ashkenazi. O diagnóstico precoce da doença pode se útil no acompanhamento e tratamento das manifestações apresentadas.
Doença de Canavan
É uma doença causada pela falta de uma enzima denominada aspartoacilase (ASPA), levando ao acúmulo do ácido N-acetilaspártico (ANA). O acúmulo de ANA leva a danos cerebrais, retardo mental e outros sintomas, que serão descritos a seguir. A maioria das crianças portadoras dessa doença parece normal ao nascimento, mas os sintomas aparecem em torno dos 3 a 5 meses de idade, observando-se incapacidade motora, retardo mental, macrocefalia (cabeça grande), convulsões, cegueira, postura anormal (braços flexionados e pernas esticadas), atonia muscular (sobretudo no pescoço), irritabilidade, entre outros.
A morte ocorre normalmente antes dos quatro anos de idade, embora haja relatos de sobrevivência até os 20 anos de idade. Apesar de ser uma doença sem cura, existem tratamentos para o alívio de sintomas. Estes incluem a terapia física e ocupacional, um tubo de alimentação em caso de dificuldade para comer e alguns medicamentos para convulsões e alívio da dor. Aproximadamente um em cada 40 judeus Ashkenazi é portador deste gene da doença, localizado no cromossomo 17.
Disautonomia Familiar
É um distúrbio neurológico caracterizado por uma desordem degenerativa progressiva que provoca dificuldade de engolir e se alimentar, episódios de vômitos, sudorese anormal, dor e insensibilidade a temperatura, diminuição da capacidade de sentir dor, incapacidade de produzir lágrimas e escoliose. É uma doença causada por mutações no gene IKBKAP, encontrado no cromossomo 9, caracterizada por uma desordem degenerativa progressiva que afeta os sistemas autonômico e sensorial, levando ao mau funcionamento destes. Um em cada 30 judeus Ashkenzim é portador do gene da doença, a qual atualmente não possui cura. O tratamento visa controlar os sintomas e evitar complicações, usando técnicas especiais de alimentação, terapias artificiais, medicamentos, lágrimas artificiais e gestão ortopédica.
Doença de Gaucher
É caracterizada pelo acúmulo de glicolipídio nas células do baço, do fígado e dos ossos, ou seja, órgãos que fazem parte do sistema imunológico do ser humano. O sintoma mais comum é fadiga crônica causada por anemia. É causada por mutações do gene da enzima glucocerebrosidase que se torna deficiente.
A doença foi classificada em três tipos:
Tipo 1: é o tipo mais comum e também conhecido como não neuropático, ou seja, que não envolve o sistema nervoso. Sua incidência entre os Ashkenazi está em cerca de 1:450 indivíduos. Os sinais e sintomas podem aparecer em qualquer idade, sendo o mais comum o aumento do baço e/ou do fígado. Também pode ocorrer dor, fraturas ósseas, hemorragia nasal e falta de energia.
Tipo 2: é a forma neuropática aguda da doença, portanto, atinge o sistema nervoso. É diagnosticada durante a infância e caracteriza-se por um início rápido e progressivo dos sintomas, em média entre 4 e 6 meses, e por um grande comprometimento neurológico. A evolução da doença é muito rápida, assemelhando-se à Doença de Tay-Sachs.
Tipo 3: é um tipo neuropático subagudo que manifesta-se em crianças e adolescentes, sendo a idade de início variável. Possui uma progressão mais lenta que a do tipo 2. O diagnóstico preciso e definitivo da doença de Gaucher pode ser feito com um simples exame de sangue que avalia a atividade da enzima glicocerebrosidase. Também pode ser feito com amostra da pele e biópsia da medula óssea. O tratamento consiste em uma forma modificada da enzima glucocerebrosidase administrada por via endovenosa.
Fibrose Cística
É uma doença que afeta tanto a população caucasiana como a população Ashkenazi, geralmente diagnosticada na infância. As crianças mais afetadas não falam, andam ou se desenvolvem além do nível de um ano de idade 1-2. A expectativa de vida pode ser normal, e não há atualmente nenhum tratame6nto eficaz.
Ela é causada por uma mutação no gene CFTR, presente no cromossomo 7, responsável pela produção do suor, mucos e sucos digestivos, afetando principalmente pulmões, trato gastrointestinal e sistema reprodutor. Nos pulmões, as secreções causam uma obstrução da passagem de ar, o que dificulta a respiração e facilita a ocorrência de infecções respiratórias. O pâncreas fica incapaz de produzir algumas enzimas importantes necessárias para a absorção e o processamento de gorduras.
Os sintomas mais comuns são: pele com um sabor salgado, tosse persistente, pneumonia, apetite excessivo, pouco ganho de peso e fezes volumosas. Portadores dessa doença podem ser diagnosticados antes do nascimento, através de um exame genético, ou no início da infância, pelo teste do suor. A expectativa de vida pode ser normal, embora a maioria dos portadores morram em torno dos 20 a 30 anos. Não há, atualmente, nenhum tratamento eficaz.
O Comitê ACOG em Genética (The American College of Obstetricians and Gynecologists) faz as seguintes sete recomendações:
1. A história familiar de pessoas que desejam a gravidez, ou que já estão grávidas, deve determinar se algum dos membros do casal é de ascendência judaica do leste Europeu (Ashkenazi) ou se há algum parente com uma ou mais das doenças genéticas listadas acima.
2. Triagem de portadores da TSD, Doença de Canavan, Fibose Cística e Disautonomia Familiar deve ser oferecida aos indivíduos judeus Ashkenazi antes de engravidarem ou durante a gravidez precoce, de modo que o casal tenha oportunidade de considerar as opções de testes de diagnóstico pré-natal. Se a mulher já está grávida, é necessário avaliar ambos os parceiros simultaneamente para que os resultados sejam obtidos em tempo hábil a assegurar que os testes de diagnóstico pré-natal sejam possíveis.
3. Indivíduos de ascendência judaica Ashkenazi devem perguntar sobre a disponibilidade de triagem de portadores de outros distúrbios. A triagem de portadores está disponível para mucolipidose IV, Doença de Niemann-Pick tipo A, Anemia de Fanconi grupo C, Síndrome de Bloom e Doença de Gaucher. Materiais educativos podem ser disponibilizados para que os pacientes interessados estejam informados para decidir sobre testes de rastreio adicionais. Alguns pacientes podem se beneficiar do aconselhamento genético.
4. Quando apenas um dos parceiros é de origem judaica Ashkenazi, ele deve ser examinado em primeiro lugar. Se ele é um portador, ao outro parceiro deve ser oferecida também a pesquisa genética (triagem). No entanto, o casal deve ser informado da baixa frequencia de portadores entre indivíduos não-judeus.
5. Aos indivíduos com história familiar positiva de uma destas perturbações, deve ser oferecida a pesquisa genética para verificar se é um portador para este distúrbio específico, pois pode se beneficiar do aconselhamento genético.
6. Quando ambos os parceiros são portadores de uma dessas doenças, eles devem ser encaminhados para o aconselhamento genético, onde será oferecido o diagnóstico pré-natal. Casais portadores devem ser informados sobre as manifestações da doença, a gravidade e as opções de tratamentos disponíveis. O diagnóstico pré-natal por “DNA-based tests” pode ser realizado em células obtidas por biópsia de vilo corial e amniocentese ou pelo PGD (Diagnóstico Pré-Implantacional) na fertilização in vitro.
7. Quando o indivíduo for portador, os seus parentes têm também o risco se serem portadores da mesma mutação. O paciente deve ser encorajado a informar seus parentes de tal risco e da disponibilidade dos testes genéticos para a identificação do gene. O médico não precisa entrar em contato com esses parentes, porque a confidencialidade com o paciente deve ser mantida.
A triagem de portadores é voluntária. Consentimento informado e garantia de confidencialidade são obrigatórios. Para todas essas desordens, um resultado negativo do teste de triagem, para um ou ambos os parceiros, reduz significativamente a possibilidade de um indivíduo afetado na prole.
Os pacientes devem considerar, antes da decisão de passar por um teste genético, a prevalência da doença, o risco de transmissão e a gravidade da mesma, além de levar em conta se poderão se submeter a um tratameto de fertilização assistida, caso o resultado seja positivo.
Bibliografia
American College of Obstetricians and Gynecologists. Preconception and Prenatal Carrier Screening for Genetic Diseases in Individuals of Eastern European Jewish Descent Number 442.. Obstet Gynecol 2009; 114:950-3.
American College of Obstetricians and Gynecologists. Screening for Tay-Sachs disease. ACOG Committee Opinion No. 318. Obstet Gynecol 2005;106:893–4.
Buchalter, M. S; Wannmacher, C. M; Wajner, M. Tay-Sachs disease: screening and prevention program in Porto Alegre. Rev. Bras. genetica;6(3):539-47, 1983.
Dong J, Edelmann L, Bajwa AM, Kornreich R, Desnick RJ. Familial dysautonomia: detection of the IKBKAP IVS20 (+6T—>C) and R696P mutations and frequencies among Ashkenazi Jews. Am J Med Genet 2002;110:253–7.
Eng CM, Desnick RJ. Experiences in molecular-based prenatal screening for Ashkenazi Jewish genetic diseases. Adv Genet 2001;44:275–96.
Geller, Mauro; Squeff, Fabiano Alves; Guerra, Juliana Elvira Herdy; Lima, Olímpia Alves Teixeira. Aspectos genéticos e clínico-terapêuticos da Doença de Tay-Sachs / Genetic and clinical therapeutics aspects of Tay-Sachs disease. J. Bras. Med. 81(5/6):17-22, nov.-dez. 2001.
Harper JC, Sengupta SB. Preimplantation genetic diagnosis: state of the art 2011. Hum Genet. 2012 Feb;131(2):175-86. doi: 10.1007/s00439-011-1056-z. Epub 2011 Jul 12. Review.
Kaback, Michael M (December 2000). “Population-based genetic screening for reproductive counseling: the Tay–Sachs disease model”. European Journal of Pediatrics 159 (Suppl 3): S192–S195.
Kingsmore S. Comprehensive carrier screening and molecular diagnostic testing for recessive childhood diseases. PLoS Curr. 2012 May 2:e4f9877ab8ffa9.
Rozenberg, Roberto e Pereira, Lygia da Veiga. The frequency of Tay-Sachs disease causing mutations in the Brazilian Jewish population justifies a carrier screening program. Sao Paulo Med. J., July 2001,vol.119, no.4, p.146-149.
“Tay–Sachs disease Information Page”. National Institute of Neurological Disorders and Stroke. 14 February 2007. Archived from the original on 29 December 2011.
Zlotogora J, Bach G, Munnich A. Molecular basis of mendelian disorders among Jews. Mol Genet Metab 2000;69:169–80
Conheça alguns outros sites de Arnaldo Schizzi Cambiaghi: Fertilidade Natural – Gravida Feliz
